
Benign atrophic papulosis – The wedge-shaped dermal necrosis can resolve with time
C.C. Zouboulis,* A. Theodoridis, M. Brunner, C.M. Magro
Departments of Dermatology, Venereology, Allergology and Immunology, Dessau Medical Center, Theodore Fontane Medical University of Brandenburg, Dessau, Germany Freiburg Veins Center, Freiburg, Germany Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, New York, NY, USA *Correspondence: C.C. Zouboulis. E-mail: christos.zouboulis@klinikum-dessau.de
Abstract
Background Atrophic papulosis is a rare thrombo-occlusive disease, characterized by the appearance of multiple atrophic porcelain-white skin papules, with a surrounding erythematous rim, which are histologically consisting of wedge-shaped necrosis of the dermis.
Objective It consists of two variants: (i) the benign atrophic papulosis (BAP) only involving the skin and (ii) the malignant atrophic papulosis (MAP) also involving several internal organs with a cumulative five-year survival rate of approx. 55%. While the probability of only having a BAP at onset is approximately 70%, increasing to 97% after 7 years of monosymptomatic cutaneous course, no close long-term follow-up of the development of the skin lesions has been reported.
Methods We present a precise visual documentation of the evolution of the disseminated skin lesions in a female patient with BAP spanning over two decades. Results A considerable improvement and/or clinical resolution of the majority of the lesions disputing the scarring character of the atrophic porcelain-white skin papules has been detected.
Conclusion BAP not only exhibits an excellent prognosis, but resolution of lesions can also occur after a considerable period of time.
Case report
Clinical picture
The 54-year-old female patient presented in November 1997 with multiple erythematous papules with atrophic porcelainwhite centre on the torso and the extremities. The development of the lesions had started some months before with the appearance of the first ones on the lower extremities and later scattering on the body and the arms (Figs 1-3). The lesions were appearing at a rate of 30–40 each month. Initially, no symptomatology was described; however, after some months, the lesions became intensively painful mainly in the foot area.
Laboratory data
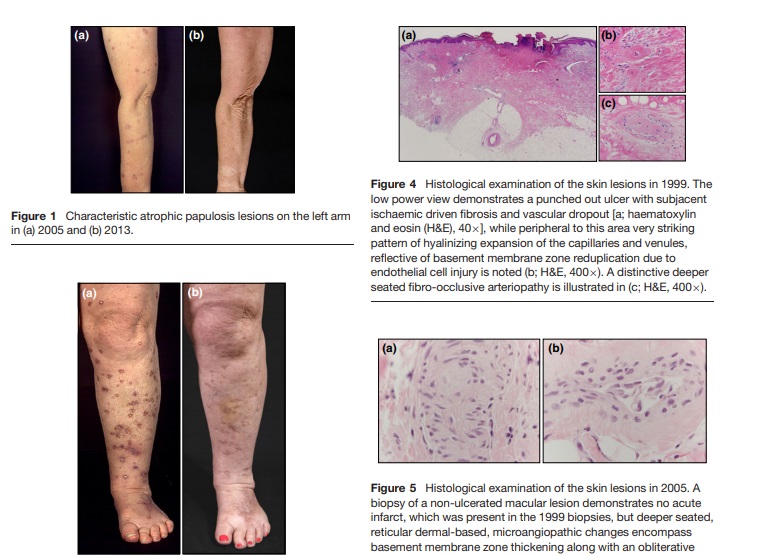
Skin biopsies performed at the central depressed ulcerated areas and non-ulcerated macules in January 1998 and in 1999 confirmed the clinical diagnosis of atrophic papulosis1,2 and demonstrated a characteristic histological constellation. In intact non-ulcerated lesional skin, there was a striking thickening of the basement membrane zone of venules and capillaries along with a fibro-occlusive arteriopathy in the deeper dermis. Some degree of angiocentric lymphocytic infiltration was observed, but overall the process was pauci-inflammatory. The overlying epidermis was attenuated. The biopsy of the depressed necrotic lesion clinically showed a punched out ulcer with severe vascular thrombosis in the zone of infarction while peripheral to the area of necrosis capillaries and venules demonstrated a significant pattern of vascular basement membrane zone reduplication with variable thrombosis along with a distinctive deeper seated fibroocclusive arteriopathy (Fig. 4a-c). There was also striking mucin deposition within the dermis and subcutaneous fat. Further laboratory analysis including blood cell count, blood sedimentation rate, c-reactive protein, serum proteins, protein electrophoresis, electrolytes, liver enzymes, bilirubin, alkaline phosphatase, creatinine, urea, uric acid, ANA, cryoglobulins, factor V Leiden, prothrombin mutation, proteins C and S, antithrombin III, fibrin monomers, lupus anticoagulant, cardiolipin antibodies, plasminogen, Quick, PTT, thrombin time, fibrin time, fibrinogen as well as D-dimers was in the normal range. A C677T mutation of the methylene tetrahydrofolic acid reductase was not detected. The platelet aggregation tests (with adenosine phosphate, collagen, adrenalin, arachidonic acid and ristocetin) were reduced responding to the acetyl salicylic acid treatment. An abdominal endoscopy revealed no specific changes.
Treatment and follow-up
The patient received initially a topical steroid therapy without any effect because the lesions continued to spread on all body parts, while many of them became extremely painful. Due to the intensity of the skin lesions, a systemic cyclosporine A therapy was administered, however, without any signs of improvement. Cyclosporine A was discontinued because of a worsening of the ulcerations of the lesions on the lower extremities with a simultaneous local infection with multiresistant staphylococci that required hospital admittance. Other therapeutic efforts with methotrexate, oral corticosteroids, intravenous immunoglobulins and topical tacrolimus failed to improve the condition. The appearance of new lesions ceased in 2006, while she was treated with oral pentoxifylline.3,4 A biopsy of a non-ulcerated macular lesion demonstrated a deep reticular dermal-based occlusive arteriopathy whereby small arteries in the 200 micron size calibre range exhibited an occlusive fibromucinous thrombus and as well other capillaries and venules showed vascular basement membrane zone thickening (Fig. 5). However, the intensity of the pain remained, which required the occasional administration of oral and intravenous analgesics (carbamazepine, gabapentin, amitriptyline and sertraline). Furthermore, due to a deep venous thrombosis of the right leg in 2006 without apparent reason, a long-term oral warfarin therapy was started.
Since 2013, the patient presents a considerable improvement and/or resolution of the majority of the atrophic porcelain-white skin papules (Figs 1-3). The current therapy consists of apixaban (following warfarin) and pentoxifylline.
Discussion
Atrophic papulosis (Kohlmeier € –Degos disease)2 consists of two variants, the benign atrophic papulosis (BAP) involving only the skin and the malignant atrophic papulosis (MAP) involving the skin and several internal organs.1 The prognosis is determined mainly by the presence of systemic involvement: the cumulative five-year survival rate in patients with systemic disease is approx. 55%, while none of the patients with only cutaneous disease has a lethal outcome.1 The probability of only having a BAP at onset is approximately 70%, increasing to 97% after 7 years of monosymptomatic cutaneous course. The age at disease onset is 36.5 years (range 5–60), not differing between BAP and MAP.1
The classical histology of atrophic porcelain-white skin papules with a surrounding erythematous rim shows a wedgeshaped connective tissue necrosis, due to thrombotic occlusion of the small arteries deep in the corium.5,6 Confocal microscopy could detect additionally densified collagen fibres and a decrease in dermal capillaries inside the connective tissue necrosis but presence of capillary hyperplasia in the periphery of the lesion.7 Magro et al.8 proposed that the striking microvascular thrombosis is attributable to vascular C5b-9 deposition, while the obliterative fibrointimal expansion may be linked to enhanced expression of type I interferons, as revealed by the extent of tissue expression of MXA, a type I interferon-inducible protein as well as endothelial tubuloreticular inclusions. In particular, it is known that the type I interferons may play a role in the peripheral blood recruitment of monocytes into the intima with subsequent myofibroblastic differentiation and as well the type I interferons exert an inhibitory effect on endothelial progenitor cell recruitment. Both of these effects could eventuate in the distinctive fibro-obliterative arteriopathy, a morphologic finding that persists despite complement inhibition.9 Harvell et al.10 examined histological specimens of the lesions according to the duration of their existence. Early lesions have shown a superficial and deep perivascular lymphocytic infiltration, with distinct mucin deposition, which can resemble tumid, nonscarring, discoid lupus erythematosus. The fully developed lesions show changes that are common to long-standing ischaemia characterized by marked epidermal thinning with subepidermal fibrosis and vascular dropout, highly reminiscent of changes seen in fibrosing variants of dermatomyositis, the prototypic autoimmune endotheliopathy syndrome, as well as lichen sclerosus et atrophicus.11 They reported these characteristics to possibly be compatible with a minimal variant of lichen sclerosus et atrophicus.
Our patient with numerous disseminated skin lesions presented a considerable improvement and/or resolution of the majority of the lesions several decades after the BAP onset. This observation may indicate that the wedge-shaped connective tissue necrosis, due to thrombotic occlusion — in the context of an acute fibrin thrombotic event or the unique fibromucinous arterial occlusion — in atrophic papulosis does not lead to a permanent scar formation and can resolve with time.